

Genomic Data Analytics
(1) Next Generation Sequencing techniques generate vast amount of genetic data that requires highly scalable and efficient tools for analyzing these data. Sequencing reads from the machines can be in the range of TB-PB range, sequential algorithms can no longer meet the needs. In our lab, we focus on developing De Bruijn graph based approach for assembling the raw reads. Through mathematical abstraction and building an asynchronous computing model, we have developed SWAP-Assembler for large scale genome assembly. We have demonstrated that SWAp-Assembler can scale to tens of thousands of cores on Mira@Argonne National Lab, and Tianhe-1A@National Supercomputing Center in Tianjin. This is an open source project, and the software can be download from http://sourceforge.net/projects/swapassembler/.
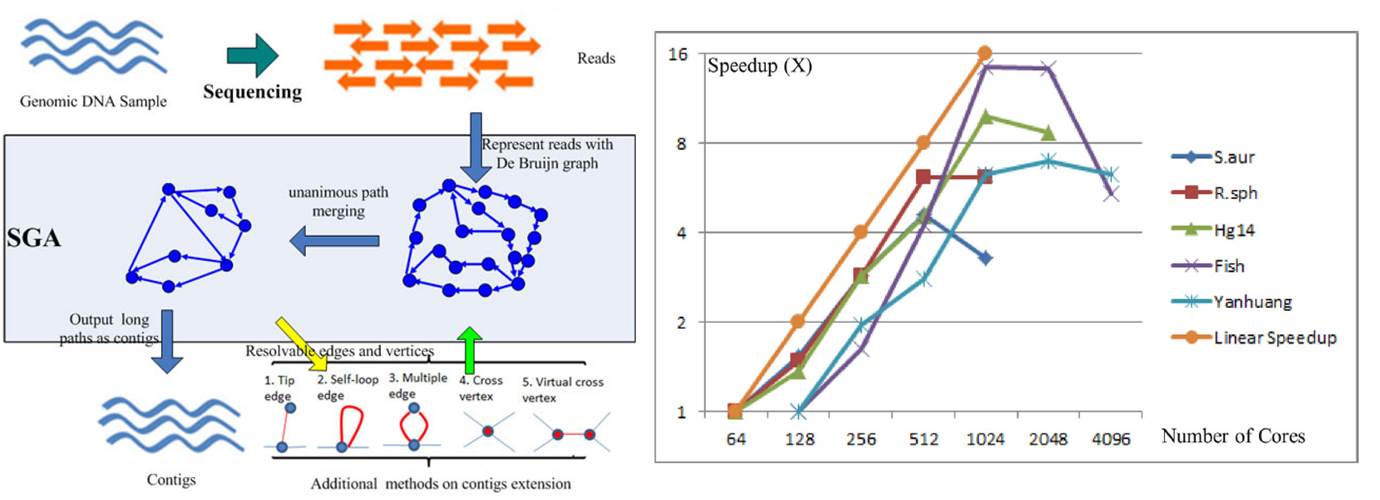 De Bruijn Graph based Genome Assembly and its High Scalability on Tianhe 1A
De Bruijn Graph based Genome Assembly and its High Scalability on Tianhe 1A
(2) Many circRNA transcriptome data were deposited in public resources, but these data show great heterogeneity. Researchers without bioinformatics skills have difficulty in investigating these invaluable data or their own data. Here, we specifically designed circMine (http://hpcc.siat.ac.cn/circmine) that provides 1 821 448 entries formed by 136 871 circRNAs, 87 diseases and 120 circRNA transcriptome datasets of 1107 samples across 31 human body sites. circMine further provides 13 online analytical functions to comprehensively investigate these datasets to evaluate the clinical and biological significance of circRNA. To improve the data applicability, each dataset was standardized and annotated with relevant clinical information. circMine provides userfriendly web interfaces to browse, search, analyze and download data freely, and submit new data for further integration, and it can be an important resource to discover significant circRNA in different diseases.
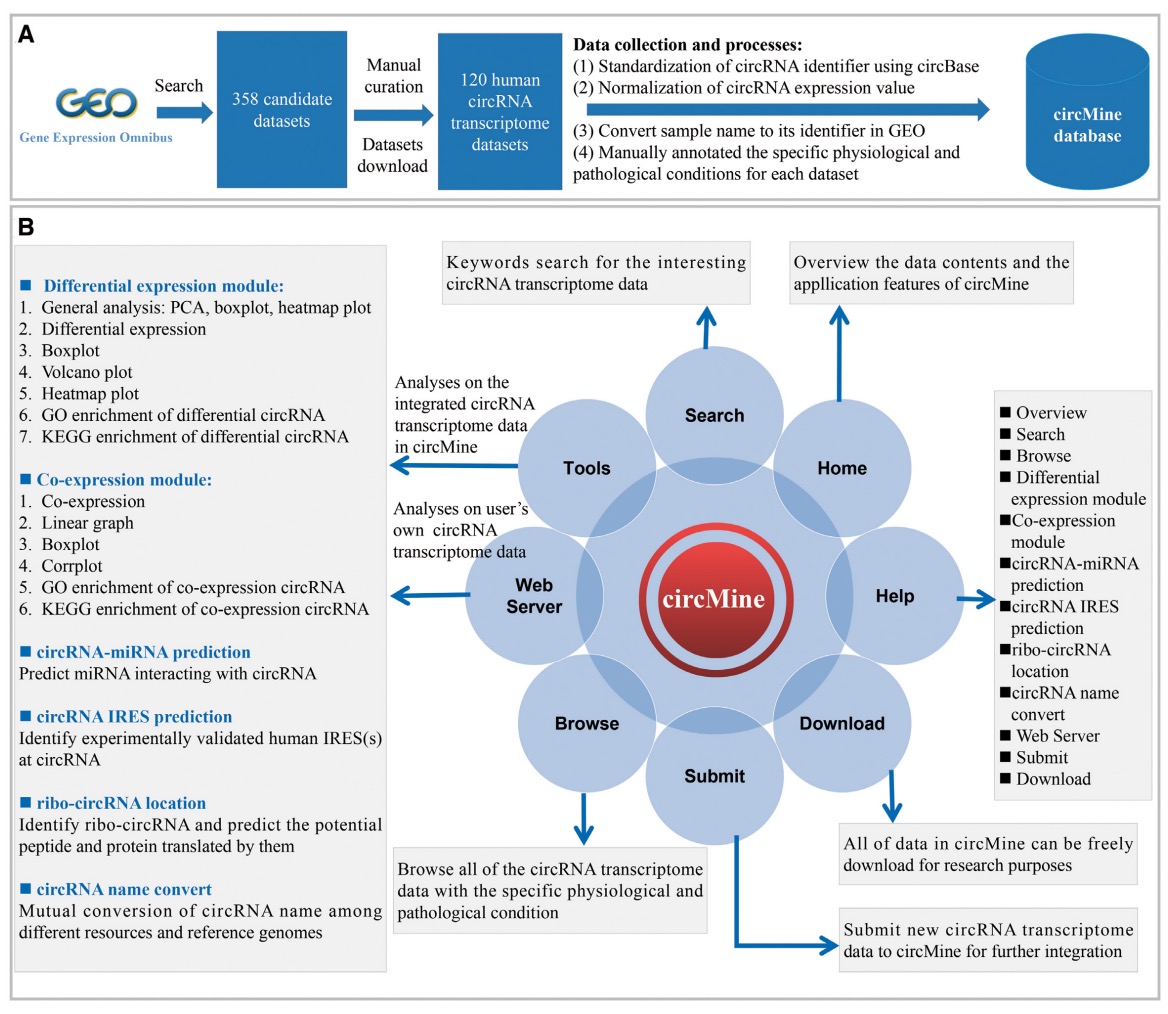 The scheme for data collection and manual curation of circMine
The scheme for data collection and manual curation of circMine