

Protein Folding & Structure Prediction
(1) We have recently proposed Replica Exchange with Tunneling (RET) to overcome some of the shortcomings of replica exchange molecular dynamics by integrating ideas from hybrid MC/MD into the replica exchange protocol. We use the RET approach to study the association of misfolded proteins into small soluble oligomer and their conversion into insoluble fibrils, processes that are difficult to probe experimentally. For a 11-residue amyloid forming cylindrin peptide we show that RET allows for a more efficient sampling of the formation and interconversion between fibril-like and barrel-like assemblies. We describe a protocol for optimized analysis of RET simulations that allows us to propose a mechanism for formation and interconversion between various cylindrin assemblies. Especially, we show that an interchain salt bridge between residues K3 and D7 is crucial for formation of the barrel structure.
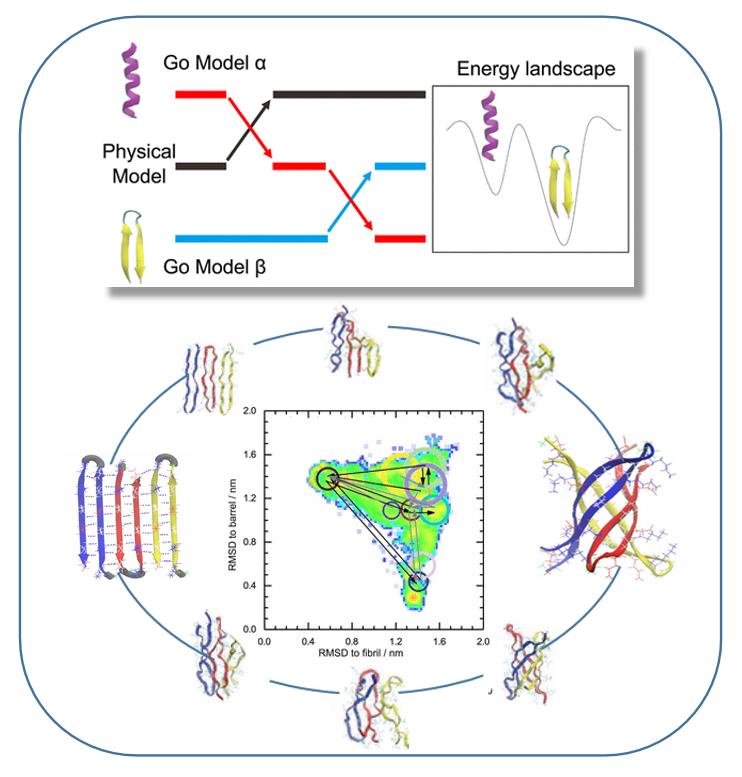
Replica Exchange with Tunneling (RET) and its application the formation and interconversion mechanism between fibril-like and barrel-like assemblies
(2) Sequence-based residue contact prediction plays a crucial role in protein structure reconstruction. In recent years, the combination of evolutionary coupling analysis (ECA) and deep learning techniques has made tremendous progress for residue contact prediction, thus a comprehensive assessment of current methods based on a large-scale benchmark data set is very needed. Relying on the high-performance computing platform, we perform a large-scale evaluation on different types of contact predictors according to a wide range of perspectives. Through a retrospective analysis of traditional machine learning /evolutionary coupling analysis methods/ consensus machine learning methods and a multi-perspective study on recently developed deep learning methods, we explore the most advanced contact predictors, pursue application scenarios for different methods, and seek prospective directions for further improvement.
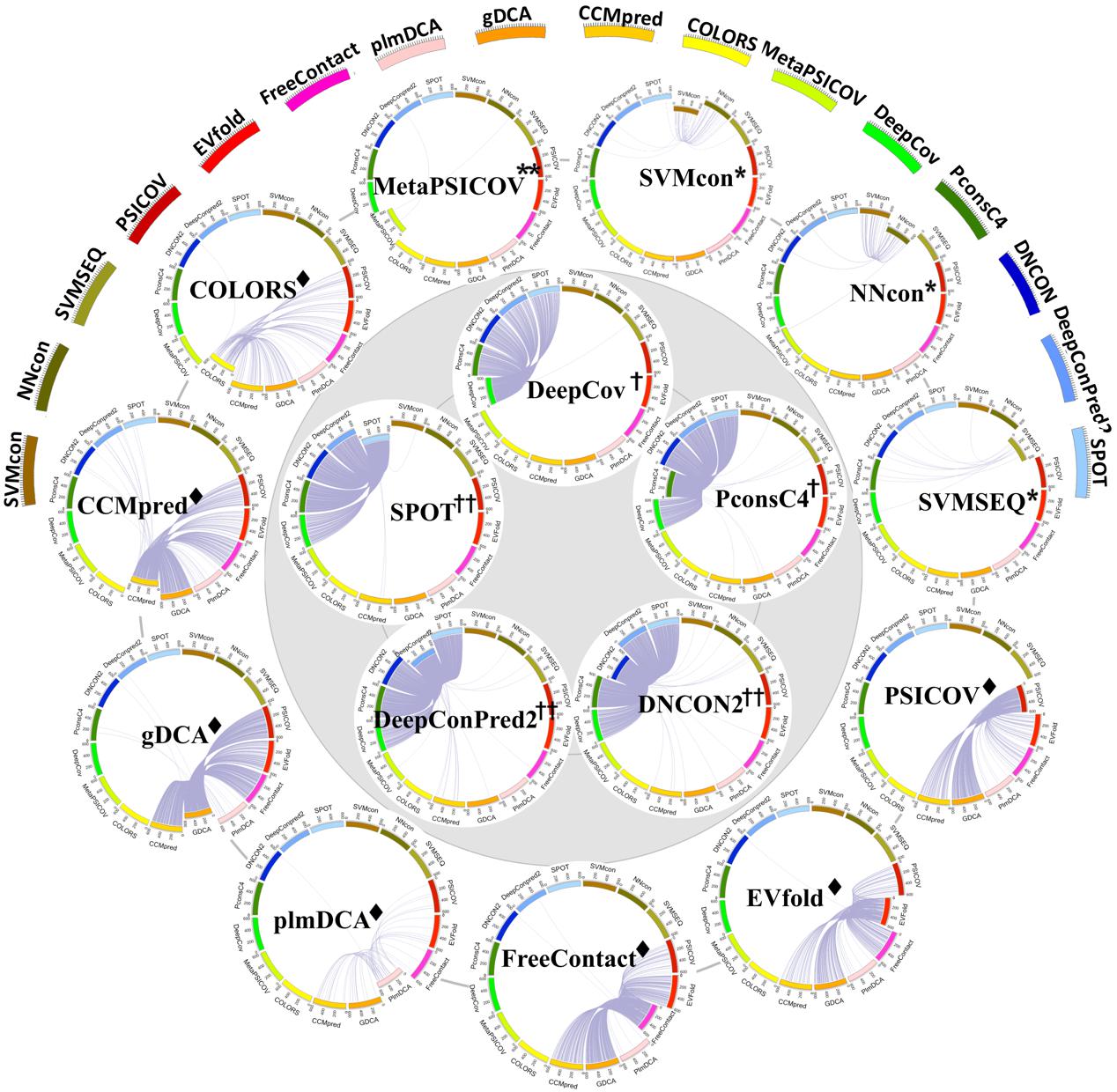 Prediction similarity evaluationof different methods based on 610 highly independent proteins
Prediction similarity evaluationof different methods based on 610 highly independent proteins